转录组概述
转录组是一系列RNA的集合(常见mRNA, lncRNA, circRNA, sRNA),推荐两篇综述阅读:A survey of best practices for RNA-seq data analysis、RNA sequencing: the teenage years.
转录组测序流程
RNA 样品检测
RNA 样品检测是建库的前置步骤,我们通过样品检测RNA样本总量与完整性,这两项指标是评判样本质量的关键点:
- Nanodrop检测RNA纯度(OD260/280)、浓度、核酸吸收峰。
- Aglient 2100检测RNA完整性(包含RIN值、28S/18S、图谱基线与5S峰)
奇奇怪怪的量都指的是啥
RIN值,即RNA Integrity Number,其反应样品的完整性如何,越接近10则样品完整性越高。
28S/18S,S即沉降系数,在真核生物的核糖体中主要含有28S、18S、5.8S与5S四种rRNA(原核生物为23S、16S和5S三种),计算28S/18S的比值(原核生物为23S/16S)可以衡量提取RNA完整性(即是否发生降解),1.8~2.0表明完整性较好,一般认为要大于等于1.5.
OD260/OD280、OD260/OD230代指吸收波长A230nm、A260nm与A280nm之间的比值,它们分别代表:
- A230nm为碳水化合物最高吸收峰波长
- A260nm为核酸最高吸收峰波长
- A280nm为蛋白质最高吸收峰波长
纯RNA样品标准为 1.7 < OD260/OD280 < 2.0(<1.7代表有蛋白或酚的污染,>2.0表明可能有异硫氰酸残留),OD260/OD230比值约为2.5(小于2.0表明存在糖类,盐类或有机溶剂的污染)。
2100峰图在集合了以上数据的基础上给出了样品检测峰图。
Aglient 2100
Agilent 2100 生物分析仪是一种“芯片上的实验室”技术,它会让片段跑过充满凝胶的微小通道(就像是电泳仪,或者说色谱仪一样),小的片段先到达,大的片段后到达,从而测出我们样品中各个大小片段的分布,一般情况下,我们的样本最好是集中在一些特定的bp长度下的。
如果发生了降解,那图谱基线就会上升(不同碱基长度的片段产生,抬升了图谱基线)。
建库
样品合格后,就需要进行建库(建立测序文库),主要流程如下:
- 用带有Oligo(dT)的磁珠富集真核生物mRNA(此步骤对RNA的完整性要求比较高,一般RIN值要大于8,测序偏差)
- 加入Fragmentation Buffer将mRNA进行随机打断。(早期技术的限制使得测序读长有限,必须限制长度)
- 以mRNA为模板,用六碱基随机引物(random hexamers)合成第一条cDNA链,然后加入缓冲液、NTPs、RNase H和DNA polymerase I 合成第二条cDNA链,利用AMPure XP beads纯化cDNA;
- 纯化的双链cDNA再进行末端修复、加A尾并连接测序接头,然后用AMPureXP beads进行片段大小选择;
- 最后通过PCR富集得到cDNA文库。(DNA更加稳定,适合测序)
文库样式如下
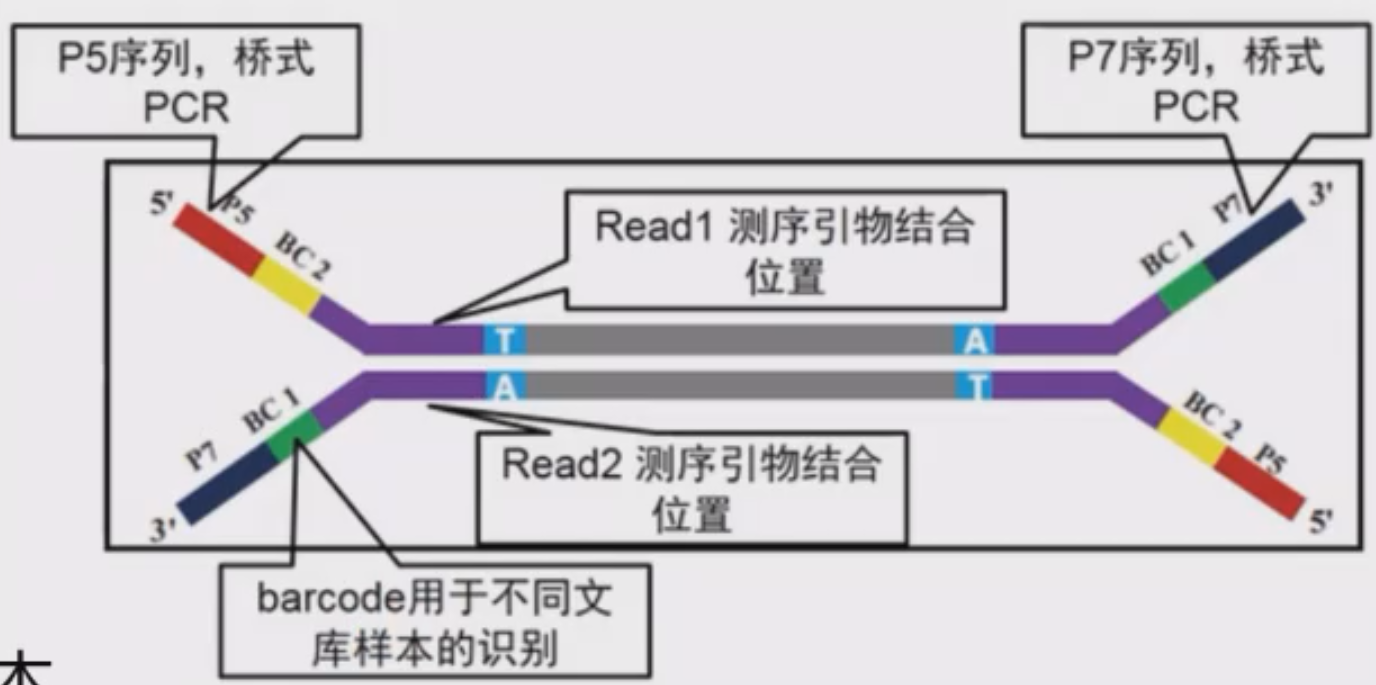
Y字接头自身不配对,用于和玻璃芯片(flow cell)上的接头连接,以进行桥式PCR。
Barcode用于唯一标识不同测序结果的样本来源(一次测序可能会混杂很多样本来源,以节省成本)。
文库中载荷包含待测序序列与测序引物结合位点。
文库质控
使用 Qubit2.0 初步定量,使用Agilent 2100对文库的insert size检测,使用Q-PCR方法对文库的有效浓度进行定量(一般认为要大于2 nMol)。
Qubit2.0
Qubit2.0是一种荧光计,特殊的染料只会在与DNA结合的情况下发光,通过定量样品荧光强度就可以估算出我们的文库总浓度。
insert size
理想状态下扩增后的cDNA片段大小应该集中在一个区间内(注意我们已经将其打碎了),通过Agilent 2100就可以测出这些文库的长度分布。
Q-PCR
通过实时荧光定量PCR测出那些已经被正确组装好的文库数量(有效文库浓度),原理是设计特定的引物,使得其只能扩增被正确组装的文库(比如直接加入测序引物)。
与此同时我们还要介绍一个文库质量评估方法——mRNA片段化随机性检验,虽然其应当在建库时对mRNA进行随机打断的时候进行,但是也属于文库质控的一部分,故放在这部分介绍。
这个技术基于两个前提:已知测序目标mRNA的序列、随机断裂的真随机性,基于此背景,期望里mRNA随机断裂后形成的reads应当均匀覆盖原先的mRNA序列,那么断裂后的reads的测序结果比对在原mRNA上的分布应当是均匀的(5'-3'中间为平坦曲线)。
那么这里会有两种因素导致曲线向着3'端聚集:
- RNA降解(5'-3')导致打断后靠近3'端的reads更多,测序结果中3'端的覆盖度更高。
- RNA降解后难以被磁珠整体捕获(磁珠一般拉住3'端),最终导致测序结果中3'端覆盖更高。
上机测序
二代测序:lllumina Novaseq平台测序(DNA簇、荧光偶联与桥式PCR)
测序原理——边合成边测序(SBS),基于单分子阵列的小型芯片(上有大量Y型配对接头)、桥式PCR反应、可逆性末段终结(dNTP上修饰有叠氮基团、切割位点与荧光基团) 。